Ready 4 Action Product Planning Service
Ready 4 Action, proprietary feasibility assessment, sets the cornerstone of a cost-effective product development plan. Ready 4 Action is the four-step process used to evaluate the viability of proposed drug products. This important tool enables clients to make the go/no-go decision regarding a given drug's market potential and clinical development feasibility.Armed with the neccessary intelligence, uncover with Ready 4 Action…


Ready 4 Action, proprietary feasibility assessment, sets the cornerstone of a cost-effective product development plan.
Ready 4 Action is the four-step process used to evaluate the viability of proposed drug products. This important tool enables clients to make the go/no-go decision regarding a given drug's market potential and clinical development feasibility.
Armed with the neccessary intelligence, uncover with Ready 4 Action — including details of key factors influencing the pharmaceutical marketplace, the regulatory status and the clinical strategy involved in development of a specific drug — clients can make well-informed drug development decisions and avoid unnecessary and potentially destructive financial risks.
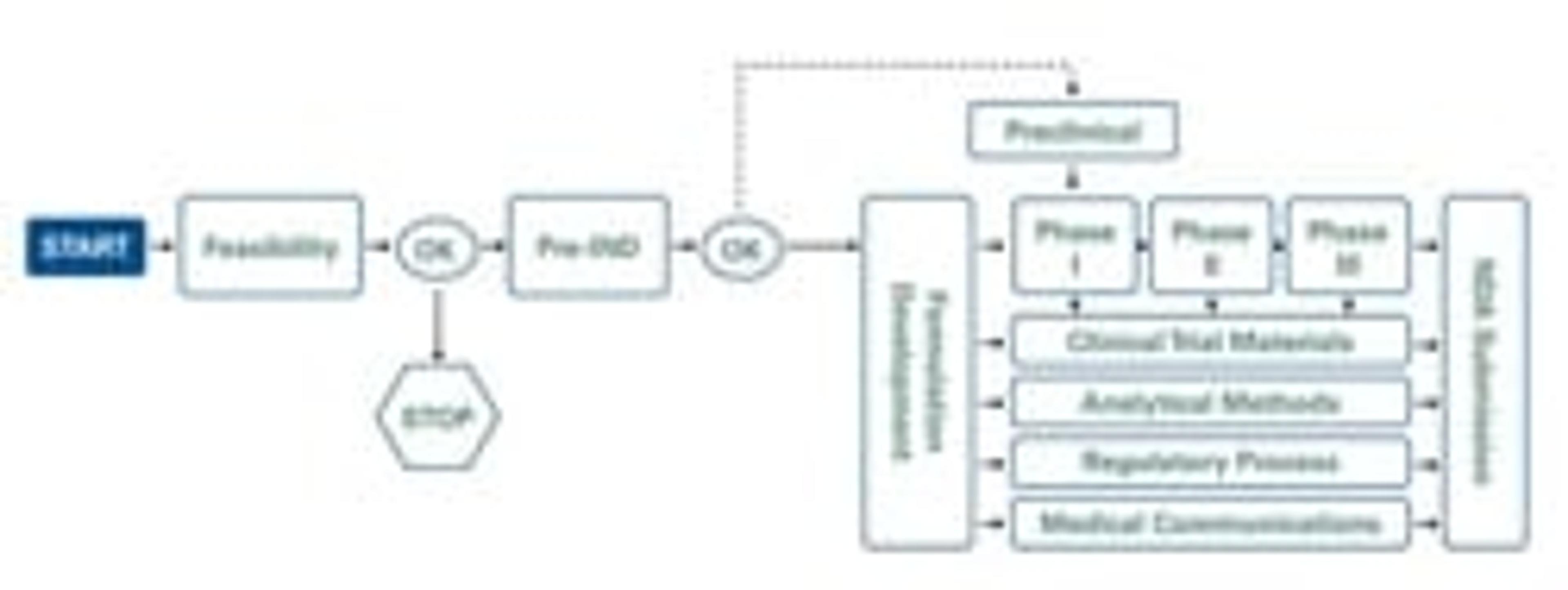
Camargo's Ready 4 Action assessment includes detailed data pertaining to your specific drug, along with a proposed clinical development plan — an invaluable feature of the assessment — and a schematic of the proposed plan. Incorporating major decision points and cost estimates, your plan includes clinical pharmacology, preclinical studies, pharmacokinetics and regulatory recommendations — that’s why it is a critical first step in the race to drug approval.
Ready 4 Action Product Planning Service Assessment:
Scientific Viability - Does the science make sense? For instance, is the formulation or chemistry practically and pragmatically achievable? Is it scalable? Are API ingredients available and affordable?
Medical Viability - Does the product have a clear niche in the medical specialty? Is it effective for solving a unique problem or solving a problem in a unique way? Does it present an acceptable risk/benefit? Is it appealing to the proposed patient population?
Regulatory Viability - What clinical trials or other data will be required to gain approval? Can development be expedited? What distinguishing information can be presented on the labeling for eventual promotional activity?
Commercial Viability - Is there a viable market for the product? What is the potential for future competition or substitution? What is needed to ensure reimbursement? What is the optimal pricing?

Unraveling the Exclusivity Determination for Prodrugs
One difficulty in bringing a prodrug to market is that the determination of market exclusivity for a prodrug has been evolving in the Food and Drug Administration (FDA) over the years. This process has included the interpretation and re-interpretation of regulations establishing what constitutes a prodrug for the purpose of defining a period of market exclusivity. If a prodrug is determined to be a new chemical entity, it is eligible for five years of market exclusivity. This white paper discusses this process, providing insight into the recent exclusivity reconsiderations for a number of previously approved prodrugs as well as knowledge about probable future decisions regarding prodrug exclusivity.
Why More Drugs than Ever are Approved Through 505(b)(2)
For many products and companies, 505(b)(2) offers a clear path to approval, a differentiated product and at least some period of marketing exclusivity. The rising tide of drugs approved under this strategy is a testament to its growing importance in the drug development market.
Identifying Products for Drug Development Programs
This white paper describes how the 505(b)(2) services offered by Camargo, can help pharmaceutical companies to identify and assist in the development of its 505(b)(2) candidate.
Understanding the 505(b)(2) Approval Pathway
A 505(b)(2) is a new drug application which contains full safety and effectiveness reports, but allows at least some of the information required for approval to come from studies not conducted by or for the applicant. This method gains approval for new drugs in a fraction of the time and cost required by traditional paths. This application note describes the features and benefits of the 505(b)(2) approval pathway.
CMC for 505(b)(2) Applications
The Chemistry, Manufacturing, and Control (CMC) Section (Modules 2 & 3 of the Common Technical Document [CTD]) of any application to the FDA will be the cornerstone for demonstrating the quality of any drug product. A clear document that complies with the ICH Harmonized Tripartite Guideline per the ICH Steering Committee Meeting (9 Nov 2000) is instrumental to that process. This poster outlines the variations that can occur from application to application depending on the dosage form, drug product, and type of file under construction, specifically a 505(b)(2) application.