How to overcome challenges with XL-MS to study whole proteomes
SelectScience speaks to the President of the German Society for Mass Spectrometry, who is using chemical cross-linking and mass spectrometry to provide deeper proteomics insights
26 Aug 2019
Editorial article

Structural mass spectrometry is being used increasingly in the field of structural biology, with one central technique being chemical cross-linking in combination with mass spectrometry (XL-MS) which is used to map protein conformations and study protein-protein interactions.
In this exclusive interview, Prof. Dr. Andrea Sinz discusses the steps her group has taken to move from single protein studies to complex networks and how they are hoping to overcome challenges with XL-MS complexity, including trying the new µPAC™ column from PharmaFluidics.
Sinz is a key scientist in this area, having worked on XL-MS since her postdoctoral degree in the United States about 20 years ago. Today, Sinz, who is President of the German Society for Mass Spectrometry, leads a research group at Martin-Luther-University Halle-Wittenberg using XL-MS to provide a deep insight into the 3D-structures of isolated protein complexes as well as protein interaction networks in cells, organisms or tissues.
What are the challenges you have with XL-MS when moving from a single protein to a complex network?
AS: I think the biggest challenge is software because every group working in the XL-MS field still has their own workflows and protocols. So now, we are working hard to establish and harmonize XL-MS workflows and software tools.
I think it is particularly important that the software is freely available, and not linked to one supplier. We have our MeroX software, and the goal is now to have a workflow that is really stable that a lot of people can apply. This will enable them to get reproducible results because, otherwise, it's very hard to judge the results from different groups.
This was the major outcome from our XL-MS harmonization study, that you see with so many protocols and the results are so diverse. Currently, when you're a beginner in that field, you don't know where to start with, and I would really like to provide a workflow that people can use and get reproducible and confident results.
So, you start with the reagent. We have developed a cleavable linker, then we have started using the µPAC™ column from PharmaFluidics as it is stable and gives you high chromatographic resolution. In the end, you will end up with more cross-link identifications. The goal is to recommend an XL-MS workflow to the community, which could involve your separation system.
What workflows are you using the µPAC™ for and what role do you see for the µPAC™ in this harmonization and the separation of these complex mixtures?
“It was easy to implement the µPAC™ column into our current systems.”
AS: We have been using the 200cm-µPAC™ column for mapping the Drosophila embryo protein interaction network. We have focused on comparing the µPAC™ column with our homemade column, which was typically a column packed with reversed-phase C18 material (3 µm, 120 Å) and a 75-micrometer inner diameter, 50-centimeter long column.
Just thinking about the Drosophila proteome, we were observing peaks in the chromatogram that could not be resolved really well. However, with the µPAC™ column I was really impressed that the whole separation was much improved, on what is a very complicated sample. For benchmarking, we analyzed 1 µg of cross-linked Escherichia coli ribosome on a 200cm-µPAC™ column with a 240-min gradient and performed a detailed comparison of cross-linked products. We found 26% more cross-linking sites and 32% more cross-link spectral matches compared to our C18 packed-bed column.
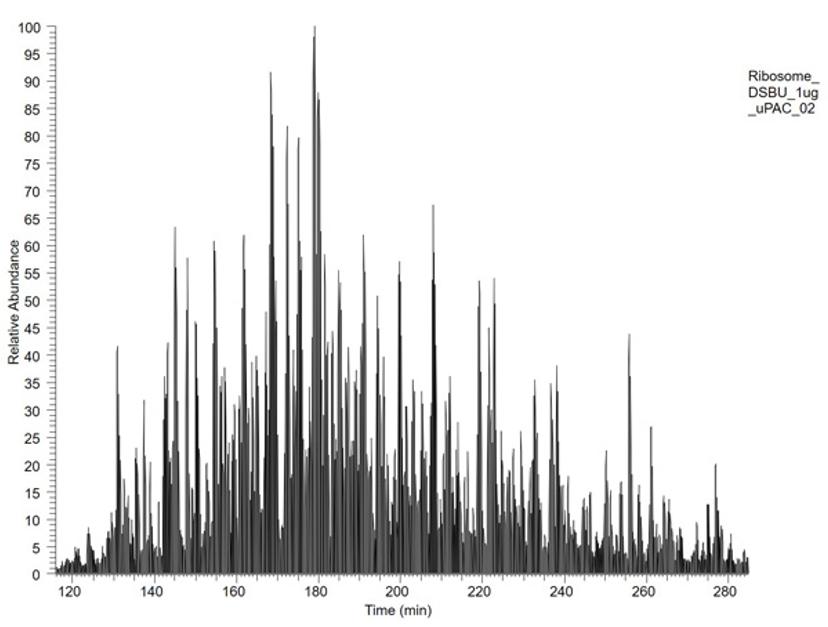
Did you see any benefits of the low back pressure for analyzing whole proteomes?
AS: The low back pressure allows the use of very long columns and our initial experiments show that increasing the LC gradient up to 480 minutes will increase even more the cross-link identifications for whole proteomes. Optimizing the LC conditions with the 200cm-µPAC™ column for the Drosophila proteome is currently underway in our lab and we are looking forward to presenting these results to the community very soon.
Do you use PharmaFluidics products in your lab? Write a review today for your chance to win a $400 Amazon gift card>>
Related products
Request Quote for All Products
50 cm µPAC™ column
PharmaFluidicsWith an internal volume of 3µL, this 50 cm long RP-C18 µPAC™ column is perfectly suited to perform high throughput analyses with shorter gradient solvent times (30, 60 and 90-minute gradients) and it can be used over a wide range of flow rates, between 100 and 2000 nL/min. µPAC™ column, 50 cm length, with pillar array backbone at interpillar distance of 2.5µm, C18.
200 cm µPAC™ column
PharmaFluidicsAs an alternative to conventional columns, PharmaFluidics offers micromachined nano-LC columns or micro Pillar Array Columns (µPAC™). These µPAC™ columns display exceptional separation properties with unprecedented peak capacity and narrow peak shapes at low operating back-pressure. µPAC™ column, 200 cm length, with pillar array backbone at interpillar distance of 2.5µm, C18.