How multiomics at ion mobility speed breaks research bottlenecks
In this guest editorial, Kelly Hines and Daniel DeBord discuss how IM-MS streamlines multiomics by capturing proteomic, lipidomic, and metabolomic information in a single, rapid analysis
16 Oct 2025
Kelly Hines, University of Georgia, and Daniel DeBord, MOBILion Systems
In the quest to understand life at its most fundamental level, scientists have long sought tools capable of revealing how proteins, lipids, metabolites, and other molecules work together to orchestrate biological complexity. Multiomics approaches have helped illuminate these relationships, yet traditional workflows often remain fragmented, requiring multiple instruments, lengthy analyses, and cumbersome data integration.
In this guest editorial, Kelly Hines, University of Georgia, and Daniel DeBord, MOBILion Systems, introduce a transformative approach: ion mobility–mass spectrometry (IM‑MS). By combining speed, resolution, and structural insight within a single experiment, IM‑MS represents a powerful step toward truly unified multiomics. This technology promises to collapse barriers between molecular classes, accelerate discovery, and provide an unprecedented window into the chemical networks that sustain life.
Deciphering complex chemical and biological systems with multiomics
Chemistry is complicated, but addressing the analytical challenge of accurately identifying and quantifying unknown compounds within complex, real-world samples is essential to understanding the world around us. Over the past 20-plus years since the term 'omics' was coined, it has come to represent a methodology for cataloging the chemistry that drives higher-level behavior in complex systems like human biology and the environment.
As every child learns in school, in order to explain a mathematical equation, all variables must be sufficiently defined. Similarly, to understand higher-level processes and systems such as genetic heredity, cell replication, or acid rain, it is necessary to first identify the critical chemical elements and compounds that contribute to the overall behavior of the system.
Analytical capabilities for the identification of unknown compounds have largely evolved over time, with a specific focus on one particular chemical class at a time. This approach has given rise to the fields of proteomics, metabolomics, lipidomics, glycomics, and transcriptomics, among others.
This type of focused analysis typically derives from the need to exploit unique chemical properties to perform the measurement. In some cases, this could entail spectroscopic interrogation of a particular chemical bond moiety, amplification of low-concentration RNA species via polymerase chain reaction, or mass measurements of characteristic peptide fragment ions to enable identification. In each case, sensitive and specific information can be accessed, but only for a limited scope of analytes.
To adequately describe complex chemical systems, it has become obvious that a myopic focus on a single class of molecules is insufficient. Researchers require a means to access the full catalog of chemical information present, hence the desire for multiomics analysis techniques. This desire for a multi-faceted perspective can be realized via two primary means. Either data from multiple analytical techniques for detecting various chemical moieties from the same sample must be integrated into a cohesive, normalized correlation analysis, or more preferably, a single analytical technique capable of registering signals from two or more classes must be employed.
Ion mobility-mass spectrometry as a multiomics enabling technology
While the acquisition of proteomic, lipidomic, and metabolomic data can be performed in parallel using multiple instruments, the biggest bottleneck in traditional multiomics workflows is the time required to integrate these datasets into an approximation of their original biological context and interpret the significance of altered pathways to the biology of the system.
Ion mobility mass spectrometry presents an opportunity to simplify this workflow by preserving the biological context of each 'omic' component and reducing data acquisition time. This multiplexing advantage is feasible because of the structural component in the separation principle of IM-MS, in which ions separate first by size-to-charge ratio in the IM and then by mass-to-charge in the MS. For complex mixtures, the combination of IM and MS can reveal distinct trendlines that are based on the inherent structural differences between the various types of analytes (Figure 1).
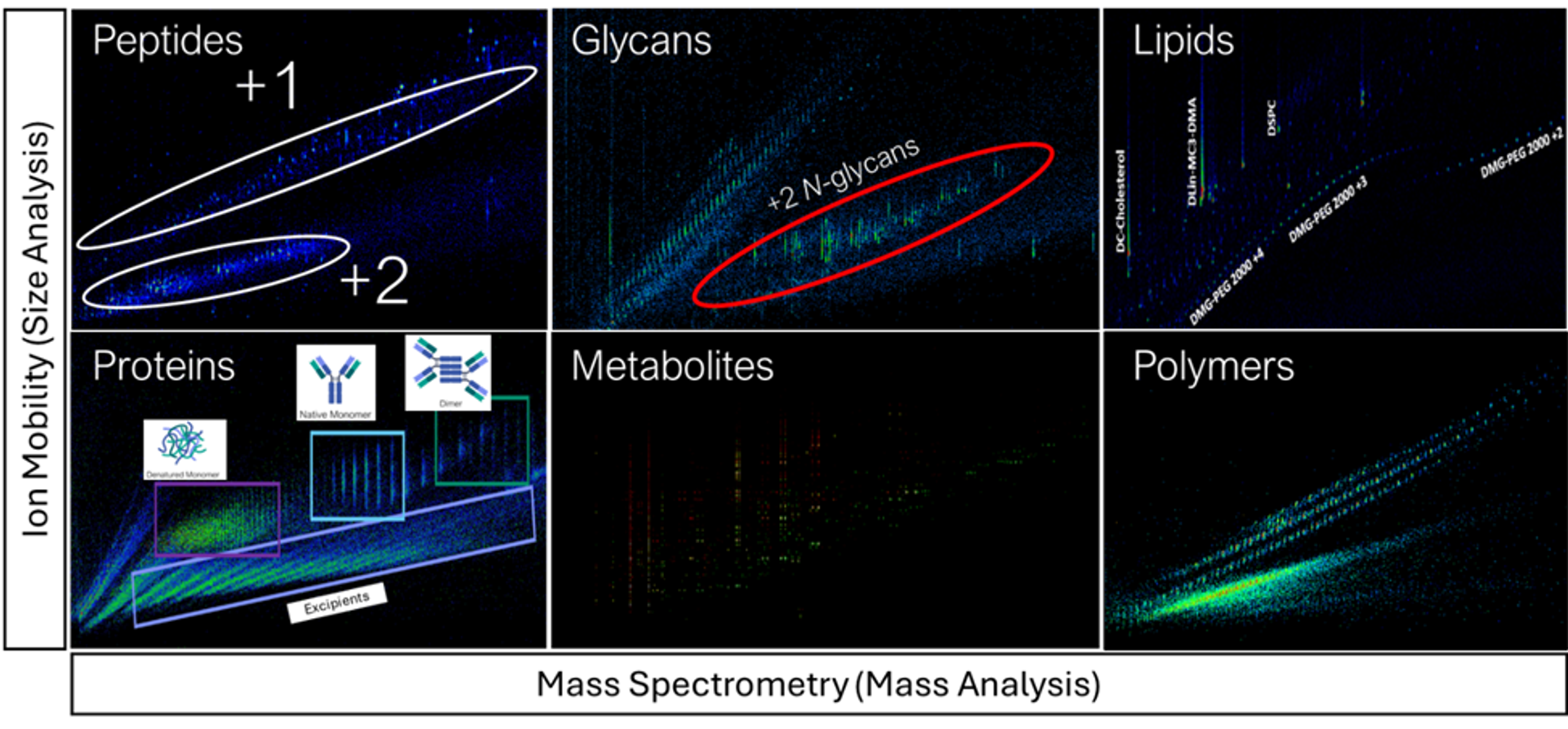
Figure 1. IM-MS heat maps demonstrating the capability of IM-MS to create characteristic trendlines across a wide range of chemical classes.
The implications for multiomics are multi-fold. The same structural moieties that distinguish lipids from metabolites and peptides can be used to resolve analytes from these classes in the gas-phase, enabling even isobaric species to be resolved in the mobility dimension(1, 2). Liquid chromatography, often used to address isobar or isomer challenges, can be reduced or eliminated entirely in favor of faster, gas-phase IM separations.
The recent availability of high-resolution IM (HRIM) instrumentation further enables the capabilities of IM for isomer separations, particularly for lipids(3) and metabolites(4), where isomers may have different biological sources or significance. Relative to liquid chromatography (LC) retention times, which can shift dramatically as columns age or due to variation in mobile phase composition, IM arrival times are inherently more stable and predictable based on gas parameters and ion physics(5).
The IM arrival times themselves are useful for the reduction or assignment of plausible identifications when transformed or calibrated to collision cross-section values. The development of predictive strategies to generate CCS values, in addition to large experimental CCS databases(6), has made it easier than ever to find(7, 8) or generate(9) a CCS value for a molecule even if no chemical standards exist.
The realization of the potential for IM-MS in multiomics analyses is not without its challenges. Ionization suppression caused by biomolecules with high ionization efficiencies is likely to outcompete other analytes if they are not first separated chromatographically. This means that, in choosing higher throughput, other analytical metrics like coverage and sensitivity may be sacrificed.
Other challenges arise from the nature of IM separations themselves, where it is possible for a single analyte to exist in two (or more) structural conformations in the gas phase. These conformers may have no biological significance but lead to an increase in the number of signals that need to be evaluated and dereplicated.
High-throughput multiomic screening? Only with IM-MS
While current multiomics workflows are geared towards discovery, IM-enabled rapid multiomics could facilitate the transition towards more targeted, high-throughput screens for the detection of specific multiomic hallmarks of disease, disease progression, or therapeutic monitoring with a single injection rather than the multi-injection, multi-instrument approach to multiomics.
The ability to perform multiomics analyses with IM-MS would enable these measurements to be performed at unprecedented throughput. Figure 2 depicts the hypothetical improvement in throughput that could be achieved with the realization of IM-MS-based multiomics. In the conventional strategy, the timescale for the complete multiomics workflow is determined by the slowest analytical steps (e.g., bottom-up proteomics) and the data integration process.
Although the sample preparation may require the same amount of time with both strategies, the IM-MS-based multiomics enables the multiplexing of the data acquisition to simultaneously acquire the metabolomic, proteomic, and lipidomic data in a single, rapid analysis. The presence of all 'omic' features within a single data file streamlines the data integration process, leading to an additional time advantage for the IM-MS-based strategy for multiomics.
This example workflow leverages pooling of the prepared samples, but alternate workflows that employ independent acquisition for each sample type still offer throughput, robustness, and data integration advantages, especially when chromatographic separations can be reduced or eliminated.
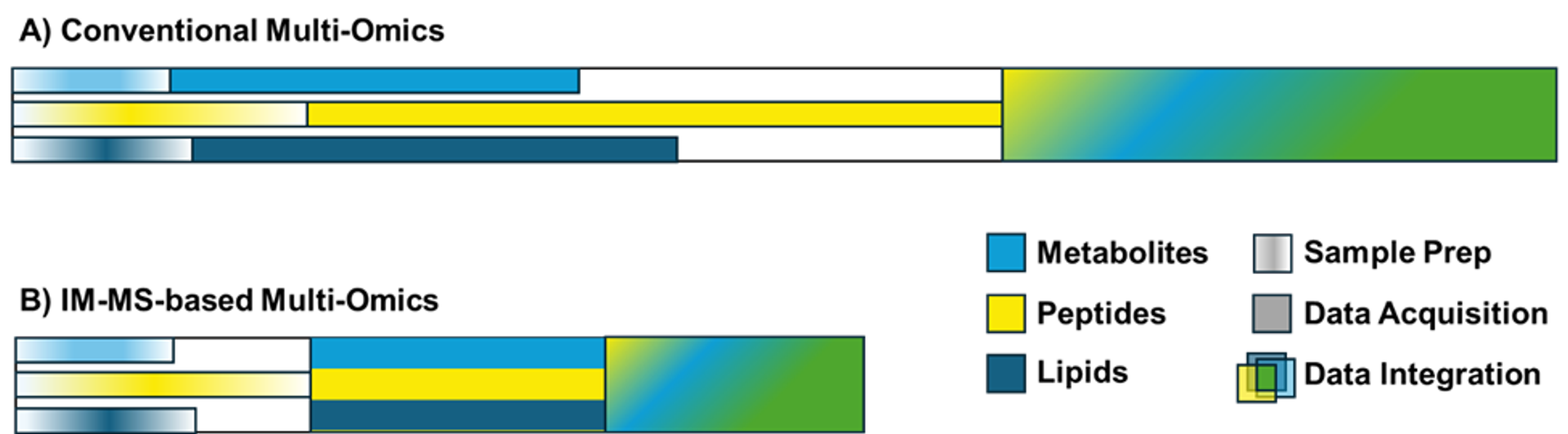
Figure 2. Timelines for sample preparation, data acquisition, and data integration for multiomics analyses performed using the conventional (A) and IM-MS-based strategies (B).
Ion mobility-mass spectrometry has the potential to transform multiomics from a fragmented, time-intensive process into a unified, high-throughput platform for understanding life’s complexity. By capturing structural and compositional diversity in a single experiment, IM-MS brings us closer to real-time molecular insights that could redefine how we study biology, disease, and the environment. As this technology continues to mature, it holds the promise of turning today’s bottlenecks into tomorrow’s breakthroughs.
References
1. L. S. Fenn, J. A. McLean, Biomolecular structural separations by ion mobility–mass spectrometry. Analytical and Bioanalytical Chemistry 391, 905–909 (2008).
2. L. S. Fenn, M. Kliman, A. Mahsut, S. R. Zhao, J. A. McLean, Characterizing ion mobility-mass spectrometry conformation space for the analysis of complex biological samples. Analytical and Bioanalytical Chemistry 394, 235–244 (2009).
3. G. Lu, S. Xu, P. Huang, L. Li, Enhancing Lipidomics With High-Resolution Ion Mobility-Mass Spectrometry. PROTEOMICS n/a, e70026 (2025).
4. X. Zheng et al., A structural examination and collision cross section database for over 500 metabolites and xenobiotics using drift tube ion mobility spectrometry. Chemical Science 8, 7724–7736 (2017).
5. S. M. Stow et al., An Interlaboratory Evaluation of Drift Tube Ion Mobility–Mass Spectrometry Collision Cross Section Measurements. Analytical Chemistry 89, 9048-9055 (2017).
6. J. A. Picache et al., Collision cross section compendium to annotate and predict multi-omic compound identities. Chemical Science 10, 983–993 (2019).
7. A. Elapavalore et al., PubChemLite Plus Collision Cross Section (CCS) Values for Enhanced Interpretation of Nontarget Environmental Data. Environmental Science & Technology Letters 12, 166–174 (2025).
8. D. S. Wishart et al., HMDB 5.0: the Human Metabolome Database for 2022. Nucleic Acids Research 50, D622–D631 (2022).
9. S. M. de Cripan et al., Predicting the Predicted: A Comparison of Machine Learning-Based Collision Cross-Section Prediction Models for Small Molecules. Analytical Chemistry 96, 9088–9096 (2024).